MetaboAnalyst is a web-based metabolomics analysis site. The majority of MetaboAnalyst’s analyses are performed using the R package.
MetaboAnalyst:https://www.metaboanalyst.ca/home.xhtml
However, there may be cases where an organisation’s security policies do not allow data to be uploaded to the web.
For this reason, the MetaboAnalyst website describes how to install the ‘MetaboAnalystR’ package, which allows similar analyses to be carried out in a local environment rather than on the web.
However, the method described causes an error when installing the ‘SSPA’ package. Information on how to solve this problem is not easy to find.
As a result, some people may want to use the MetaboAnalyst in their local environment, but find it difficult to implement.
Therefore, we will show you how to create the ‘MetaboAnalystR’ package, which is very useful if you have a specific analysis method in MetaboAnalyst, as even the R commands that MetaboAnalyst outputs can be routinely executed once you have them.
The example shows a new installation of R version 4.3.1.
Preparation
The following is the preparation required to install the “MetaboAnalystR” package: Download and install RTools from the link provided for your version of R.
RTools:https://cran.r-project.org/bin/windows/Rtools/
Next, download “SSPA_2.22.1.tar.gz” to your desktop from the link below. To download, click on the link next to “Source Package” at the bottom of the page.
SSPA_2.22.1:https://www.bioconductor.org/packages/3.8/bioc/html/SSPA.html
Finally, run the following command in R. Run the commented out part if necessary.
#Download and run RTools for your version of R
#https://cran.r-project.org/bin/windows/Rtools/
#Install devtools package
install.packages("devtools", type = "source")
#Install BiocManager package
install.packages("BiocManager")
#Install ellipse package
install.packages("ellipse")
#Install rjson package
install.packages("rjson")
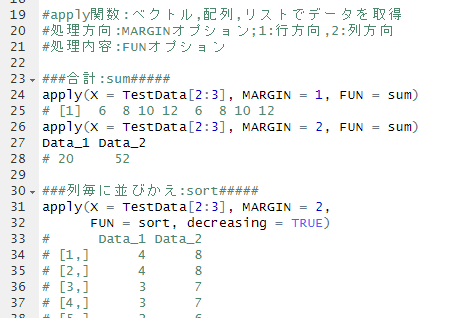
#Install OptiLCMS package
devtools::install_github("xia-lab/OptiLCMS", build = TRUE, build_vignettes = FALSE, build_manual =TRUE)
#rlang installation: Ver1.0.5 or higher required
#install.packages("rlang")
#cli installation: Ver 3.3.0 or higher required
#install.packages("cli")
#Download SSPA_2.22.1.tar.gz
#https://www.bioconductor.org/packages/3.8/bioc/html/SSPA.html
#Install required packages
BiocManager::install(c("qvalue", "limma"))
#Install SSPA_2.22.1.tar.gz
install.packages(file.choose(), repos = NULL, type = "source")Install the necessary packages in the “MetaboAanalyst” package
Run the following command.
#Preparation
metanr_packages <- function(){
metr_pkgs <- c("impute", "pcaMethods", "globaltest", "GlobalAncova", "Rgraphviz", "preprocessCore", "genefilter", "SSPA", "sva", "limma", "KEGGgraph", "siggenes","BiocParallel", "MSnbase", "multtest", "RBGL", "edgeR", "fgsea", "devtools", "crmn")
list_installed <- installed.packages()
new_pkgs <- subset(metr_pkgs, !(metr_pkgs %in% list_installed[, "Package"]))
if(length(new_pkgs)!=0){if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(new_pkgs)
print(c(new_pkgs, " packages added..."))
}
if((length(new_pkgs)<1)){
print("No new packages added...")
}
}
#Install
metanr_packages()Install “MetaboAanalyst” package
Run the following command.
devtools::install_github("xia-lab/MetaboAnalystR",
build = TRUE, build_vignettes = FALSE)If the “MetaboAanalyst” package is installed correctly, a message will appear when the package is loaded.
library("MetaboAnalystR")
MetaboAnalystR 4.0.0 initialized Successfully !
https://github.com/xia-lab/MetaboAnalystRExample
This is an example of an analysis using “cow_diet.csv” data from “Analysis Path”, “Statistics [one factor]” and “Concentration Table” from the Data Format on the MetaboAnalyst website.
and Biomarker Analysis” and “cow_diet.csv” data from the “Concentration Table”.
Data Formats:https://www.metaboanalyst.ca/docs/Format.xhtml
Note that the following commands include commands that have been analysed and output by MetaboAnalyst.
#Load locally saved "cow_diet.csv" data
FilePath <- file.choose()
#Loading the library
library("MetaboAnalystR")
#The following is the content analysed and output from the MetaboAnalyst site
#Can be run locally
# PID of current job: 1182864
mSet<-InitDataObjects("conc", "stat", FALSE)
mSet<-Read.TextData(mSet, FilePath, "rowu", "disc")
mSet<-SanityCheckData(mSet)
mSet<-ReplaceMin(mSet);
mSet<-PreparePrenormData(mSet)
mSet<-Normalization(mSet, "NULL", "LogNorm", "NULL", ratio=FALSE, ratioNum=20)
mSet<-PlotNormSummary(mSet, "norm_0_", "png", 72, width=NA)
mSet<-PlotSampleNormSummary(mSet, "snorm_0_", "png", 72, width=NA)
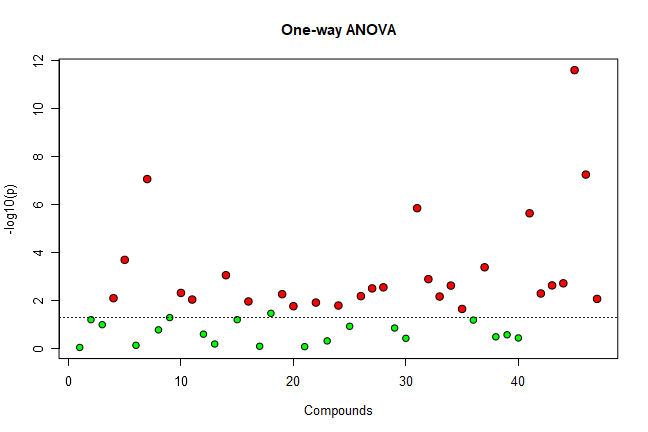
mSet<-ANOVA.Anal(mSet, F, 0.05, "fisher", FALSE)
mSet<-PCA.Anal(mSet)
mSet<-PlotPCAPairSummary(mSet, "pca_pair_0_", "png", 72, width=NA, 5)
mSet<-PlotPCAScree(mSet, "pca_scree_0_", "png", 72, width=NA, 5)
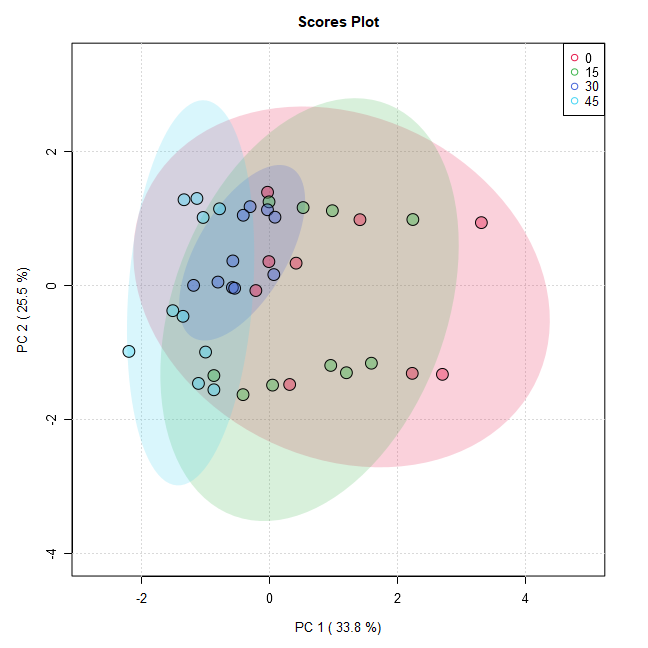
mSet<-PlotPCA2DScore(mSet, "pca_score2d_0_", "png", 72, width=NA, 1,2,0.95,0,0)
mSet<-PlotPCALoading(mSet, "pca_loading_0_", "png", 72, width=NA, 1,2);
mSet<-PlotPCABiplot(mSet, "pca_biplot_0_", "png", 72, width=NA, 1,2)
mSet<-PlotPCA3DLoading(mSet, "pca_loading3d_0_", "json", 1,2,3)Output Example
・PlotPCA2DScore command
・ANOVA.Anal command
I hope this makes your analysis a little easier !!