Rとオミクス解析:オントロジー、ネットワーク解析が出来る統合解析「dnetパッケージ」
ゲノミクスやメタボロミクスなどの生化学分野のみならず、ウェブ解析にもオミクス解析が必要ではと感じています。個人的にはオミクス解析は「1つの事柄(反応)のみではなく、事象として表現(現れる)結果を現時点で使用できる技術を最大限に生かし丸ごと解析しよう!そして新しい解釈を導きだそう」を目的とし、ビックデータは「結果を導きだす要因を探そう!データは多いほど正解に近い要因を発見できるよね?もしかしたら、気づいていない要因があるかも?」と考えています。だいぶ乱暴ですがアプローチの仕方がことなると考えています。そんなオミクス解析が出来るdnetパッケージを紹介します。データの準備が大変ですが、参考URLの例をこなすことで使えるようになると思います。
<おすすめのRに関する書籍です>

R/RStudioでやさしく学ぶプログラミングとデータ分析【ダウンロード特典付き!】 | 掌田津耶乃
Amazonで掌田津耶乃のR/RStudioでやさしく学ぶプログラミングとデータ分析【ダウンロード特典付き!】。アマゾンならポイント還元本が多数。
「dnet」パッケージの導入
下記コードを実行することで導入することができます。
参考URL: http://supfam.org/dnet/
source("http://bioconductor.org/biocLite.R")
biocLite(c("hexbin","ape","supraHex","graph","Rgraphviz","igraph","Biobase","limma","survival","foreach","doMC"))
install.packages("dnet",repos="http://cran.r-project.org",type="source")スポンサーリンク
ネットワーク図の描写
オミクス解析で良く描写される、関係性を示すネットワーク図の描写コードを示します。参照URL先には他にも例があるのでぜひご覧ください。データの準備が大変ですが、オミクス解析は非常に価値がある手法だと考えます。ぜひ、dnetパッケージを使用してみてください。
#ライブラリの読み込み
library(dnet)
#データの読み込みと設定
data(Fang)
data <- Fang
fdata <- as.data.frame(Fang.geneinfo[, 2:3])
rownames(fdata) <- Fang.geneinfo[, 1]
pdata <- as.data.frame(Fang.sampleinfo[, 2:3])
rownames(pdata) <- Fang.sampleinfo[, 1]
#解析に必要な遺伝子発現データオブジェクトの作成
colmatch <- match(rownames(pdata), colnames(data))
rowmatch <- match(rownames(fdata), rownames(data))
data <- data[rowmatch,c olmatch]
eset <- new("ExpressionSet", exprs = as.matrix(data), phenoData = as(pdata,"AnnotatedDataFrame"), featureData = as(fdata," AnnotatedDataFrame"))
#遺伝子発現値の調整
prob2gene <- function(eset){
fdat <- fData(eset)
tmp <- as.matrix(unique(fdat[c("EntrezGene", "Symbol")]))
forder <- tmp[order(as.numeric(tmp[, 1])),]
forder <- forder[!is.na(forder[, 1]),]
rownames(forder) <- forder[, 2]
system.time({
dat <- exprs(eset)
edat <- matrix(data = NA, nrow = nrow(forder), ncol = ncol(dat))
for (i in 1:nrow(forder)){
ind <- which(fdat$EntrezGene == as.numeric(forder[i, 1]))
if (length(ind) == 1){
edat[i,] <- dat[ind,]
}else{
edat[i,] <- apply(dat[ind, ], 2, mean)
}
}
})
rownames(edat) <- rownames(forder)
colnames(edat) <- rownames(pData(eset))
esetGene <- new('ExpressionSet', exprs = data.frame(edat), phenoData = as(pData(eset), "AnnotatedDataFrame"), featureData = as(data.frame(forder), "AnnotatedDataFrame"))
return(esetGene)
}
esetGene <- prob2gene(eset)
esetGene
#igraph用のデータ作成
org.Hs.string <- dRDataLoader(RData = 'org.Hs.string')
ind <- match(V(org.Hs.string)$symbol, rownames(esetGene))
#遺伝子発現値データの整形
esetGeneSub <- esetGene[ind[!is.na(ind)],]
esetGeneSub
#ネットワークデータの作成
nodes_mapped <- V(org.Hs.string)$name[!is.na(ind)]
network <- dNetInduce(g = org.Hs.string, nodes_query = nodes_mapped,
knn = 0, remove.loops = T, largest.comp = T)
V(network)$name <- V(network)$symbol
#ネットワーク解析データの準備,処理に時間がかかります。
D <- as.matrix(exprs(esetGene))
D <- D - as.matrix(apply(D,1,mean),ncol=1)[,rep(1,ncol(D))]
fdr <- dSVDsignif(data=D, num.eigen=NULL, pval.eigen=1e-2, signif="fdr", orient.permutation="row", num.permutation=200, fdr.procedure="stepup", verbose=T)
g <- dNetPipeline(g=network, pval=fdr, method="customised", nsize=30)
#ネットワーク図の作成準備
glayout <- layout.fruchterman.reingold(g)
#図書式の設定
com <- spinglass.community(g, spins=25)
com$csize <- sapply(1:length(com),function(x) sum(com$membership==x))
vgroups <- com$membership
colormap <- "yellow-darkorange"
palette.name <- visColormap(colormap=colormap)
mcolors <- palette.name(length(com))
vcolors <- mcolors[vgroups]
com$significance <- dCommSignif(g, com)
#ノードの設定
vdegrees <- igraph::degree(g)
tmp <- data.frame(ind=1:vcount(g), vgroups, vdegrees)
ordering <- tmp[order(vgroups,vdegrees),]$ind
#図のプロット
visNetArc(g, ordering=ordering, labels=V(g)$geneSymbol, vertex.label.color=vcolors, vertex.color=vcolors, vertex.frame.color=vcolors, vertex.size=log(vdegrees)+0.1, vertex.label.cex=0.4)
ordering <- tmp[order(vgroups,vdegrees),]$ind出力例
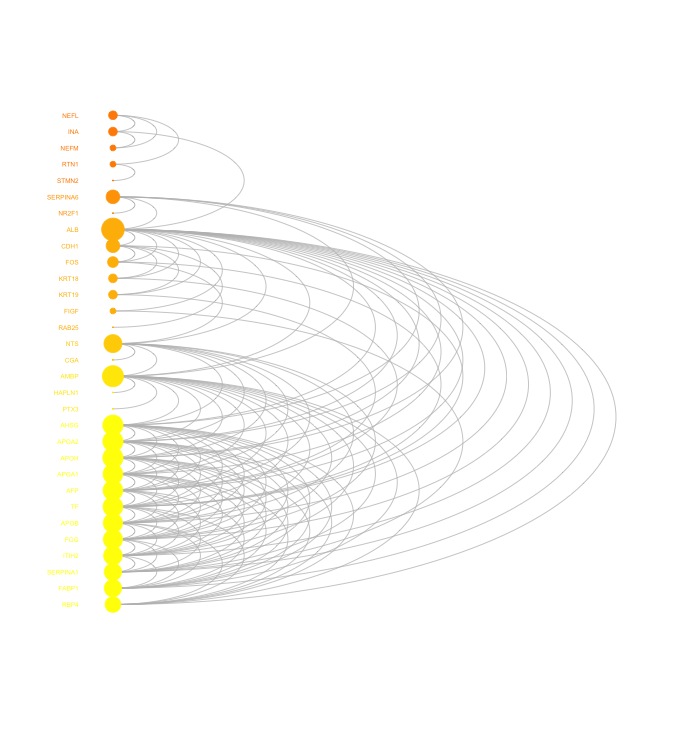
少しでも、ウェブや実験の解析が楽になりますように!!

スポンサーリンク