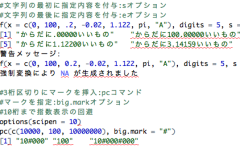
多重比較で有意水準を調整していないt検定の結果で主張している内容を見かけます。FDRをBenjamini-Hochbergなどできちんと制御しましょう。
本パッケージではBenjamini-Hochberg,Storey_adjp,Bon-EVの方法が利用できます。コマンドのコードも合わせて紹介しますので参考にしてください。
パッケージバージョンは1.0。実行コマンドはR version 3.2.3で確認しています。
パッケージのインストール
下記、コマンドを実行してください。
#パッケージの利用には「qvalue」パッケージが必要です
install.packages("BiocManager")
BiocManager::install("qvalue")
#パッケージのインストール
install.packages("BonEV")実行コマンド
詳細はコメント、パッケージのヘルプを確認してください。
#パッケージの読み込み
library("qvalue")
library("BonEV")
###仮想のp値を用意#####
set.seed(1111)
n <- 5
PValue <- sample(runif(n*10, min = 0, max = 1), n, replace = TRUE)
#####
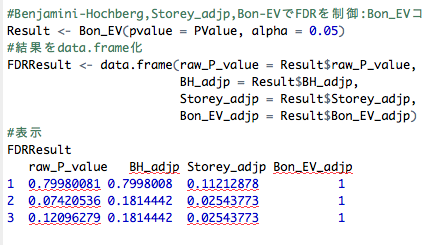
#Benjamini-Hochberg,Storey_adjp,Bon-EVでFDRを制御:Bon_EVコマンド
Result <- Bon_EV(pvalue = PValue, alpha = 0.05)
#結果をdata.frame化
FDRResult <- data.frame(raw_P_value = Result$raw_P_value,
BH_adjp = Result$BH_adjp,
Storey_adjp = Result$Storey_adjp,
Bon_EV_adjp = Result$Bon_EV_adjp)
#表示
FDRResult
raw_P_value BH_adjp Storey_adjp Bon_EV_adjp
1 0.79980081 0.7998008 0.11212878 1
2 0.07420536 0.1814442 0.02543773 1
3 0.12096279 0.1814442 0.02543773 1
#おまけ:Bon_EVコマンドのコード
#statパッケージ:p.adjustコマンド
#qvalueパッケージ:Storey JDらの方法
#論文:
#A direct approach to false discovery rates. Journal of the Royal Statistical Society, Series B, 64: 479-498.
#False discovery rates. In International Encyclopedia of Statistical Science.
#http://genomine.org/papers/Storey_FDR_2011.pdf
#Bon_EV_adjpはコードを参照
function (pvalue, alpha)
{
new_MTP_adjp <- rep(length(pvalue))
BH <- p.adjust(pvalue, "BH")
qobj <- qvalue(p = pvalue)
qvalues <- qobj$qvalues
ngene <- length(pvalue)
for (i in 1:ngene) {
adjpv <- ngene * (pi0est(pvalue)$pi0)/sum(BH <= alpha,
na.rm = TRUE) * pvalue
new_MTP_adjp[i] <- min(adjpv[i], 1)
}
mylist <- list(raw_P_value = pvalue, BH_adjp = BH, Storey_adjp = qvalues,
Bon_EV_adjp = new_MTP_adjp)
return(mylist)
}少しでも、あなたの解析が楽になりますように!!